¹³C-NMR is one of the most informative experiments in the spectroscopist's toolkit. Where ¹H-NMR tells you about the hydrogen skeleton, carbon NMR gives you the full carbon framework — quaternary centres, carbonyls, aromatic systems — all of it resolved into individual signals. The challenge is assigning those signals: connecting each peak in the spectrum to a specific carbon in the molecule.
This guide walks through a complete assignment of a real spectrum using the MID ¹³C-NMR Peak Identifier, signal by signal.
The molecule
The compound used in this example is 4'-methylacetophenone — a simple aryl ketone with a methyl group para to the carbonyl.
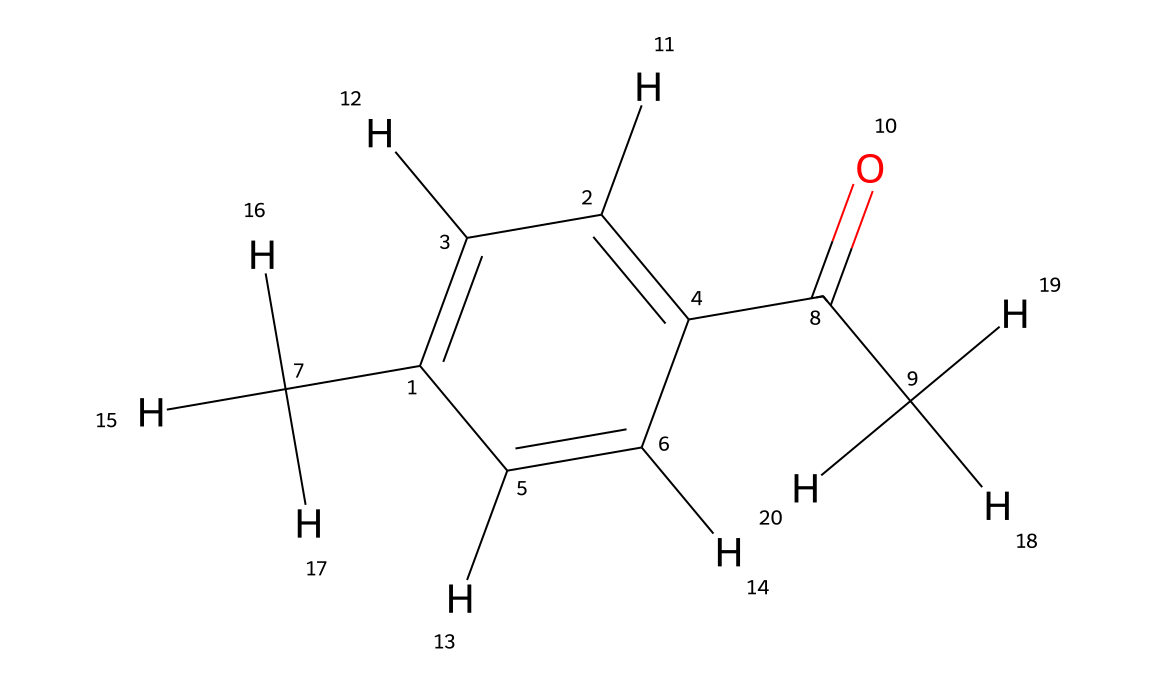
The molecule has nine carbons in total, but the para-substituted benzene ring has a mirror plane that makes C2 equivalent to C6, and C3 equivalent to C5. This reduces nine chemically distinct carbons to six observable signals in the ¹³C spectrum:
| Signal | Carbons | Environment |
|---|---|---|
| ~208 ppm | C8 | Ketone carbonyl (C=O) |
| ~149 ppm | C1 | Quaternary aromatic, ipso to methyl |
| ~142 ppm | C4 | Quaternary aromatic, ipso to C=O |
| ~136 ppm | C2, C3, C5, C6 | Aromatic CH (four equivalent carbons) |
| ~30 ppm | C9 | Methyl of acetyl group |
| ~22 ppm | C7 | Methyl on ring |
The spectrum
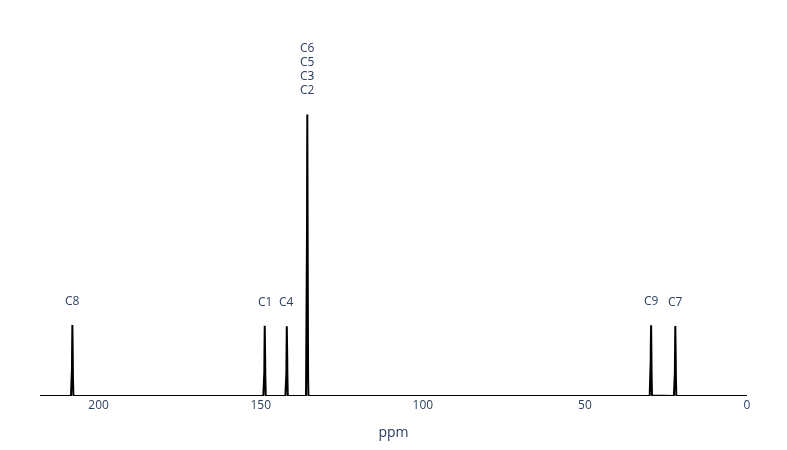
The spectrum confirms the six-signal pattern. Three things are immediately apparent. First, the isolated peak at ~208 ppm is characteristic of a carbonyl carbon. Second, the very tall peak around 135 ppm results from four carbons (C2, C3, C5, C6) all resonating at essentially the same frequency — overlapping into a single intense signal. Third, the two peaks in the aliphatic region (20–35 ppm) correspond to the two methyl groups.
With the expected signals identified, let us run each one through the peak identifier.
Using the ¹³C-NMR Peak Identifier
The Peak Identifier works by comparing each entered chemical shift against a database of quantum-chemically calculated environments. For each signal, it returns a ranked list of the most probable carbon environments — represented as small structural fragments showing the connectivity around the carbon in question (the red sphere). The percentages reflect the probability that the entered shift matches each environment.
208.09 ppm — The ketone carbonyl (C8)
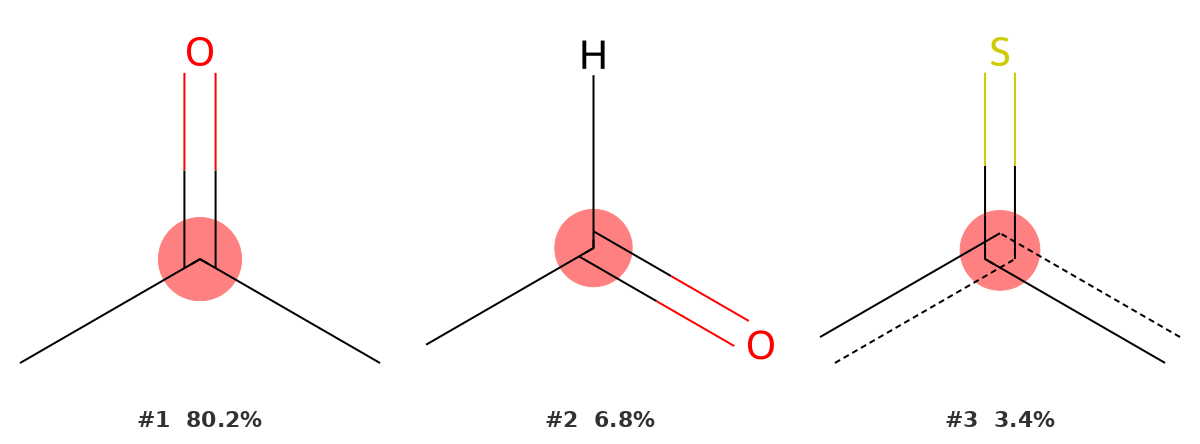
A signal at 208 ppm can only mean one thing in an organic molecule: a carbonyl carbon. The identifier assigns it to a C=O with two carbon substituents (a ketone) with a probability of 80.2% — a decisive result. The fragment shown is a carbon double-bonded to oxygen with two carbon branches, exactly matching C8 in our structure.
The gap between the top result (80.2%) and the second (6.8%) is large, which reflects how diagnostically distinctive this chemical shift region is. No other functional group resonates at 208 ppm. In practice, any signal in the 190–215 ppm range should be assigned to a carbonyl without hesitation; the tool confirms this quantitatively.
135.62 ppm — Aromatic CH carbons (C2, C3, C5, C6)
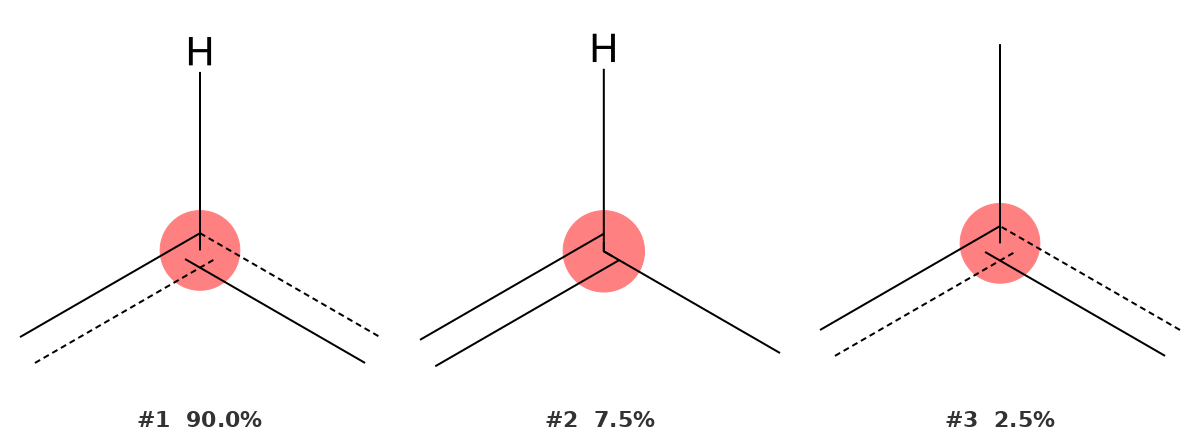
This is the tallest peak in the spectrum, and the identifier handles it with the highest confidence of any signal in this example: 90.0% probability for an aromatic carbon bearing one hydrogen. The fragment shows a trigonal carbon with one H and two bonds to the aromatic ring — precisely the environment of C2, C3, C5, and C6.
The 10% gap covered by alternatives is largely accounted for by other aromatic C–H environments at adjacent shifts. The result is unambiguous: this peak belongs to the ring CH carbons.
That this single signal represents four carbons — not one — is not something the tool needs to determine. The peak identifier assigns the type of environment; the multiplicity of carbons contributing to that signal is inferred from the molecular structure and the intensity of the peak relative to others in the spectrum.
148.75 ppm — Quaternary aromatic carbon ipso to methyl (C1)
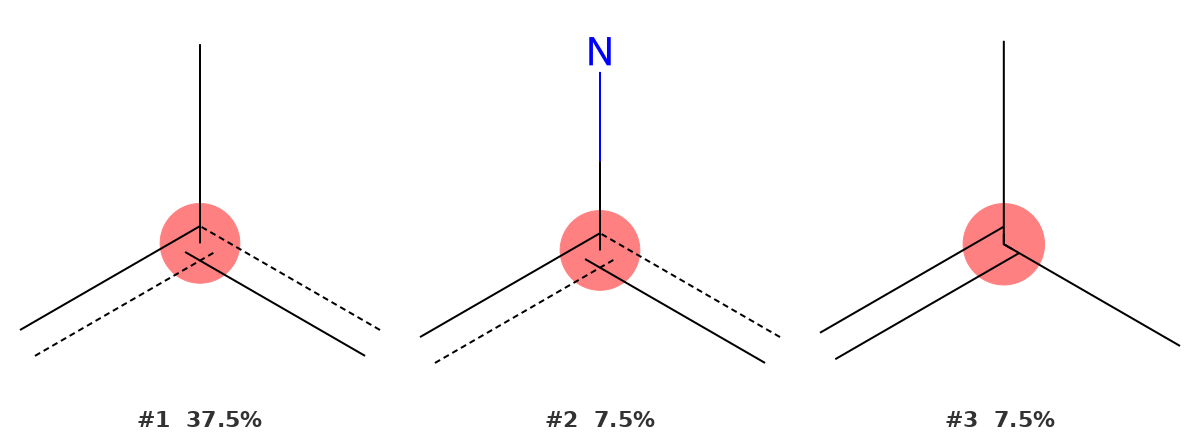
This signal belongs to C1 — the ring carbon at the point of attachment of the methyl group. It is a quaternary carbon: no hydrogen attached, three bonds to the rest of the aromatic system.
The top result is a quaternary aromatic carbon at 37.5% probability. The overall probability is lower than for the carbonyl and aromatic CH signals, which is expected. Quaternary aromatic carbons appear in a broad range of chemical shift environments (roughly 120–160 ppm), and several heteroatom-substituted variants also fall in this region, spreading probability across multiple candidates. Despite the lower absolute percentage, the correct environment still ranks first — and the structural fragment shown (trigonal carbon with three carbon bonds and no H) matches C1 exactly.
This is a good illustration of how to interpret the tool's output: rank matters more than absolute probability. When the correct environment is at the top of the list, the assignment is confirmed regardless of whether the probability is 90% or 37%.
141.94 ppm — Quaternary aromatic carbon ipso to carbonyl (C4)
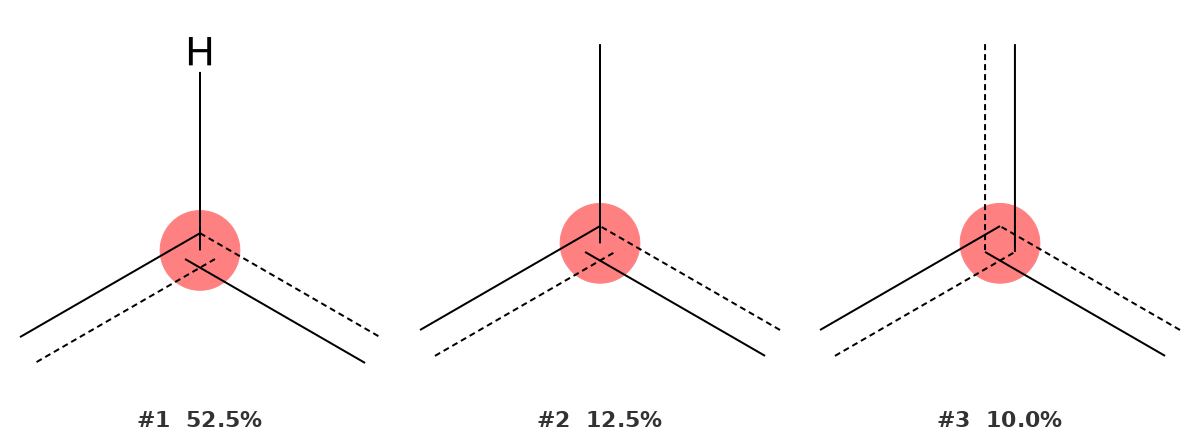
This is the most instructive signal in the example, because the top-ranked result is not the correct assignment. The identifier places an aromatic C–H first at 52.5%, while the correct environment — a quaternary aromatic carbon — appears second at 12.5%.
Why does this happen? C4, which bears the electron-withdrawing carbonyl group, resonates at 141.94 ppm — a shift that falls squarely in the region where both substituted aromatic C–H carbons and quaternary aromatic carbons appear. The tool cannot know a priori that there is no hydrogen on this carbon; it only sees the chemical shift. Because aromatic C–H carbons are statistically more common than quaternary aromatic carbons in that shift window, the C–H environment ranks first.
This is precisely the situation where prior knowledge of the molecular structure guides interpretation. We know from the structure that C4 is quaternary (it connects to C8, C3, and C5). We therefore select the second-ranked result, which matches correctly.
This case demonstrates the intended workflow: the peak identifier is not used in isolation but in combination with the known or proposed molecular structure. The tool narrows the field of possibilities; the chemist makes the final call.
22.1 ppm — Methyl on the ring (C7)
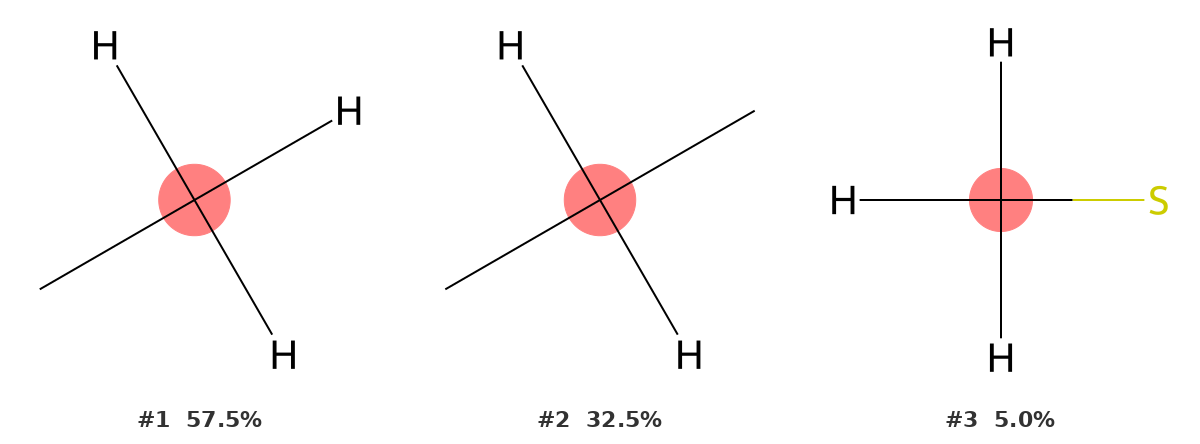
C7 is the methyl group attached directly to the aromatic ring. The identifier returns CH3 as the most probable environment at 57.5%, with CH2 second at 32.5%. The fragment for the top result shows a carbon with three hydrogen atoms and one carbon bond — a methyl group.
The second-ranked CH2 environment is not unreasonable: aliphatic methyls and methylenes partially overlap in this shift range (~15–30 ppm). However, the methyl interpretation is consistently preferred, and it is correct. In context — knowing the molecular formula and structure — there is no ambiguity.
29.59 ppm — Methyl of the acetyl group (C9)
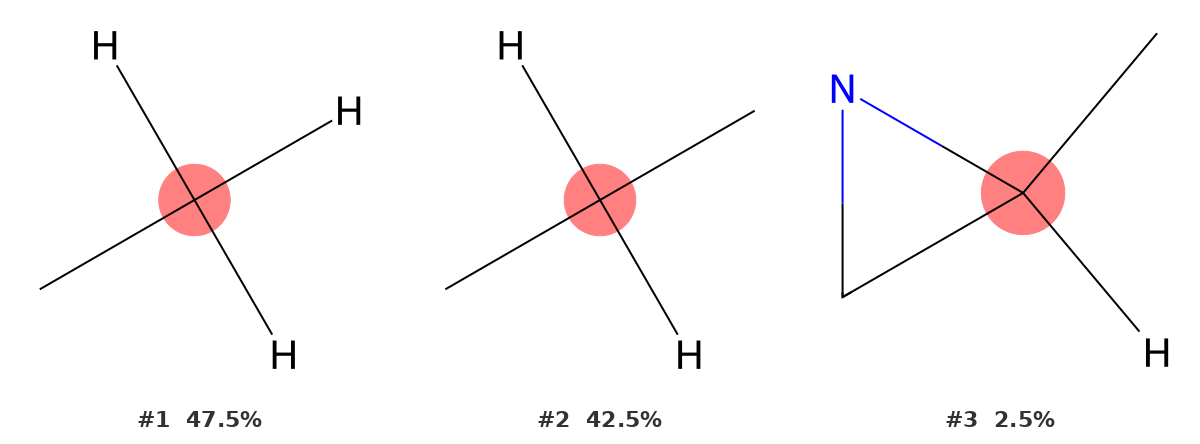
C9 is the methyl group of the acetyl substituent, directly bonded to the carbonyl carbon. It resonates slightly downfield of the ring methyl (29.59 vs 22.1 ppm), reflecting the deshielding effect of the adjacent C=O.
The identifier returns CH3 as the top result at 47.5%, with CH2 close behind at 42.5%. The two environments are genuinely similar in shift, so the close probabilities are expected. The CH3 result is correct: C9 has three hydrogens and one carbon bond (to C8).
Note that both aliphatic methyl signals in this molecule (C7 and C9) are correctly identified by the same environment type (CH3), even though they appear at meaningfully different chemical shifts. This is because both are sp³ CH3 carbons — their different shifts arise from their different positions relative to the electron-withdrawing and ring-current effects of nearby groups, not from any difference in their immediate bonding environment.
Summary
The table below collects all six assignments:
| Shift (ppm) | Carbon | Identifier rank | Probability | Correct? |
|---|---|---|---|---|
| 208.09 | C8 — ketone C=O | #1 | 80.2% | ✓ |
| 148.75 | C1 — quaternary aromatic (ipso CH₃) | #1 | 37.5% | ✓ |
| 141.94 | C4 — quaternary aromatic (ipso C=O) | #2 | 12.5% | ✓ (second result) |
| 135.62 | C2/C3/C5/C6 — aromatic CH | #1 | 90.0% | ✓ |
| 29.59 | C9 — acetyl CH₃ | #1 | 47.5% | ✓ |
| 22.1 | C7 — ring CH₃ | #1 | 57.5% | ✓ |
Five of six signals are correctly identified by the top-ranked environment. The sixth (141.94 ppm, C4) is correctly identified by the second-ranked environment — a result that is immediately obvious once the molecular structure is considered.
The complete assignment is unambiguous and consistent with the proposed structure. All six signals are accounted for, the environments match, and no unexplained peaks remain.
Conclusions
The ¹³C-NMR Peak Identifier handles all major carbon types: carbonyls, quaternary aromatic carbons, aromatic CH carbons, and aliphatic methyls. The workflow is straightforward — enter each chemical shift, read the ranked environments, and match them against your proposed structure. In most cases the correct assignment is the top result. Where it is not, structural context resolves any ambiguity quickly.
Try it with your own spectra at moleculeidentifier.com/cnmr_LO.